1. Introduction
Deep learning has gained huge success in a wide range of applications, such as computer vision, computer games, natural language processing, self-driving cars. Even more, recent advances in deep learning have made significant contributions to biological sciences and drug discovery. Many previous studies suggested deep learning techniques has shown superior performance over other machine learning algorithms in virtual screening, which is a critical step for accelerating drug discovery. However, the application of deep learning techniques in drug discovery and chemical biology are hindered due to the data availability, data processing, and the difficulties of using deep learning methods. We aim to construct a user-friendly web server for constructing deep learning models using public dataset or user provided dataset, which would help biologists and chemists virtual screening the chemical probes or drugs for a specific target of interest, or target fishing for small molecules.
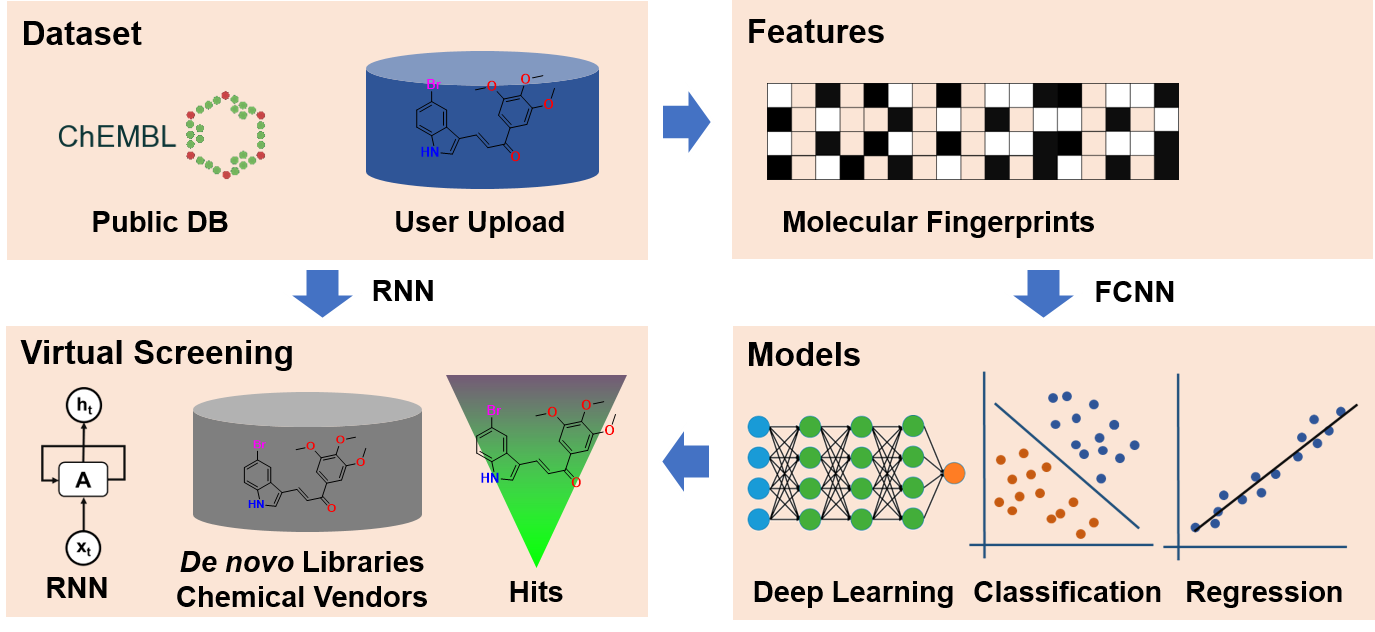
2. Framework of DeepScreening
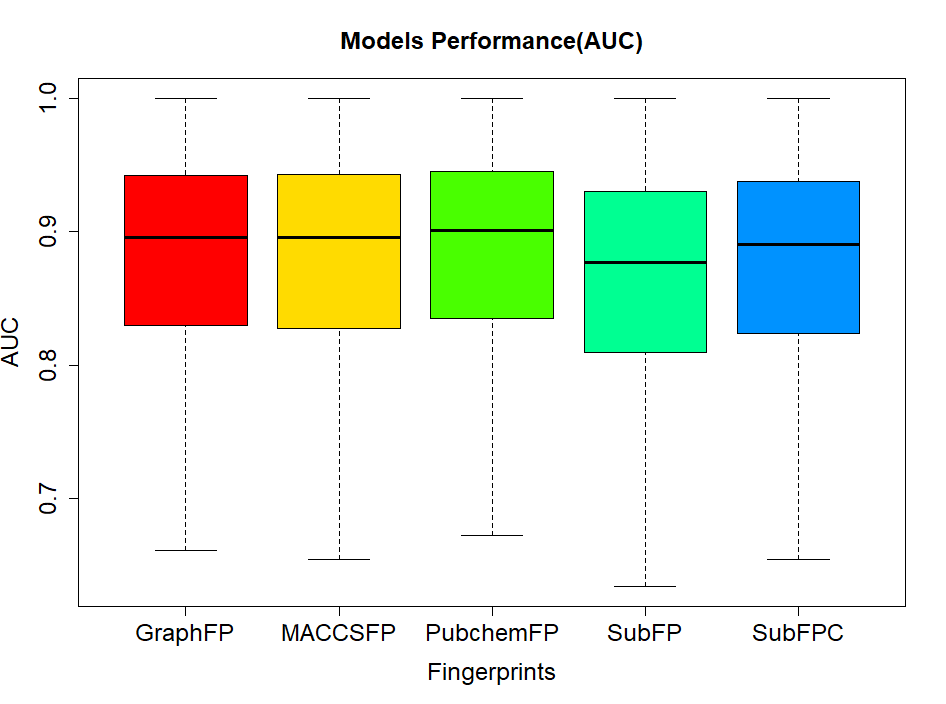
3. Models performance
In order to evaluate the screening performance of the deep learning models, we select five types of fingerprints and use the recommanded parameters against almost 966 targets dataset. The AUC values are calculated and can be ploted in following figure. The average and median of AUC is 0.86, and 0.89, which indicates a good screening performance of theose constructed deep learning models.
4. Tools used in developing DeepScreening
| # | Tools | Purpose | Link |
|---|---|---|---|
| 1 | ChEMBL24 | target and ligands source | www.ebi.ac.uk/chembl |
| 2 | ChemDoodle Web | structure draw and 3D display | web.chemdoodle.com |
| 3 | PaDEL | fingerprints and properties calculation | padel.nus.edu.sg/software/padeldescriptor |
| 4 | Pytorch | Deep learning package | pytorch.org |
| 5 | MySQL | storage database | www.mysql.com |
| 6 | Golang | web server language | golang.org |
| 7 | sklearn | model performance metrics calculation | scikit-learn.org |
| 8 | Echarts | chart visulization | echarts.baidu.com |
| 9 | bootstrap | web server language | getbootstrap.com |
| 10 | jquery | data interaction between front end and backend | jquery.com |
5. Fingerprints available in DeepScreening

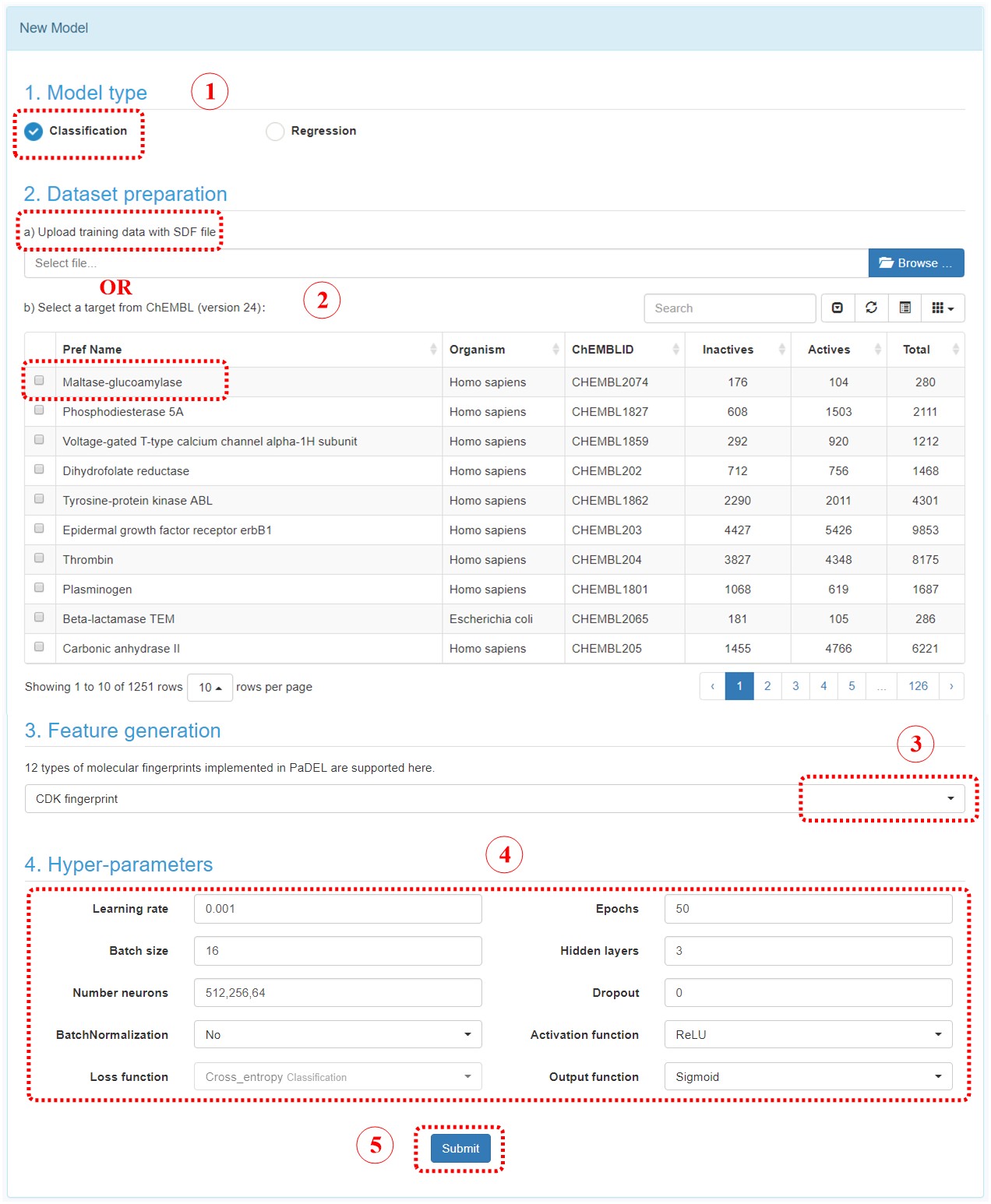
6. Steps to construct a deep learning model.
Five steps to construct a deep learning model in DeepScreening model page.
Step 1: Select a model type. Either classification or regression.
Step 2: Upload a SDF file with bioactivity information or select a target in ChEMBL. The uploaded SDF file must contain a field named “Activity”. For classification model, the label should be provided in this field, such as 0 stands for inactive, or 1 stands for active. For regression model, the bioactivity data should be provided with a numeric value, which stands for potency in nanomolar unit. User should convert other unit to the standard nanomolar unit before the SDF files are uploaded.
Step 3: Feature generation. At present stage, 12 types of fingerprints from PaDEL are provided. The bits and descriptions of those fingerprints are listed below.
Step 4: Select hyperparameters. User can select the parameters for constructing the deep neuron network. User can change the parameters and evaluate the performance of models, and find the optimum model.
Step 5: Submit.Click the submit button, which will bring you to “my model” section.
7. Functions in “my model” page.
After submitting a model, user can browse all the models in an interactive table with searching function. In the operation filed, user can update job status. When the job is finished with a status sign of “Success”, user can browse the model details, screen by using current model, and download the model to local computer.
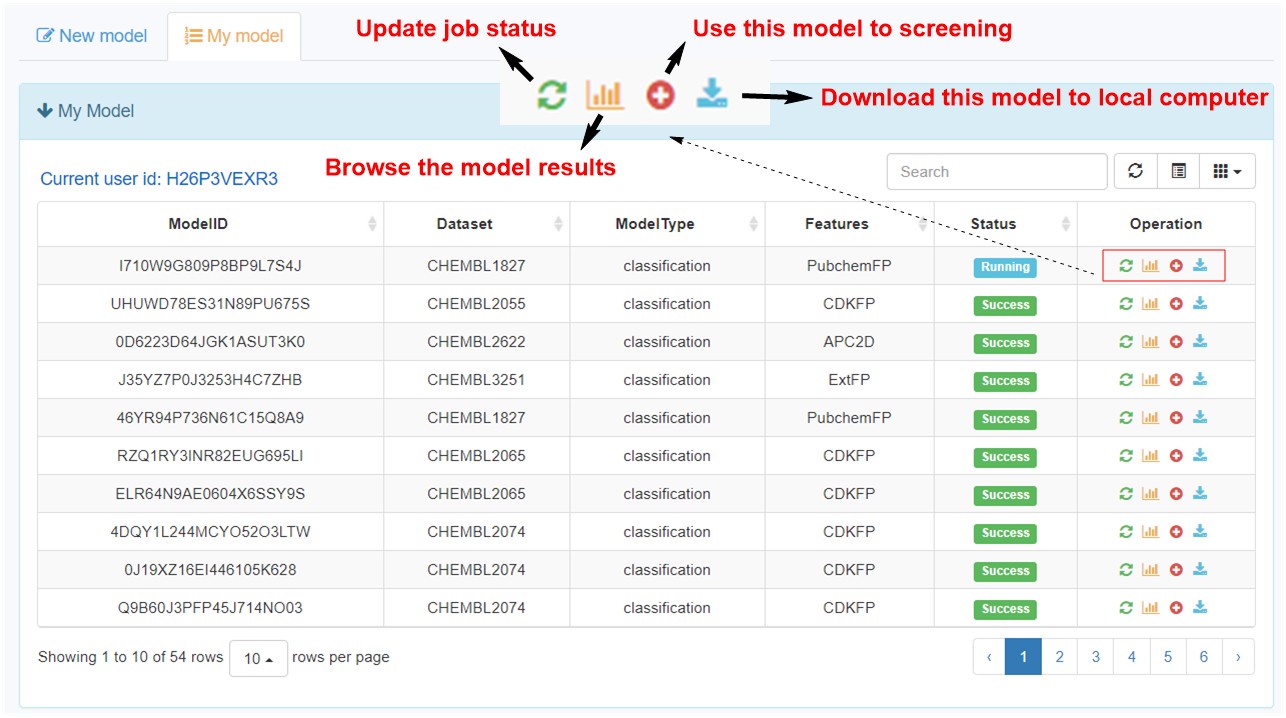
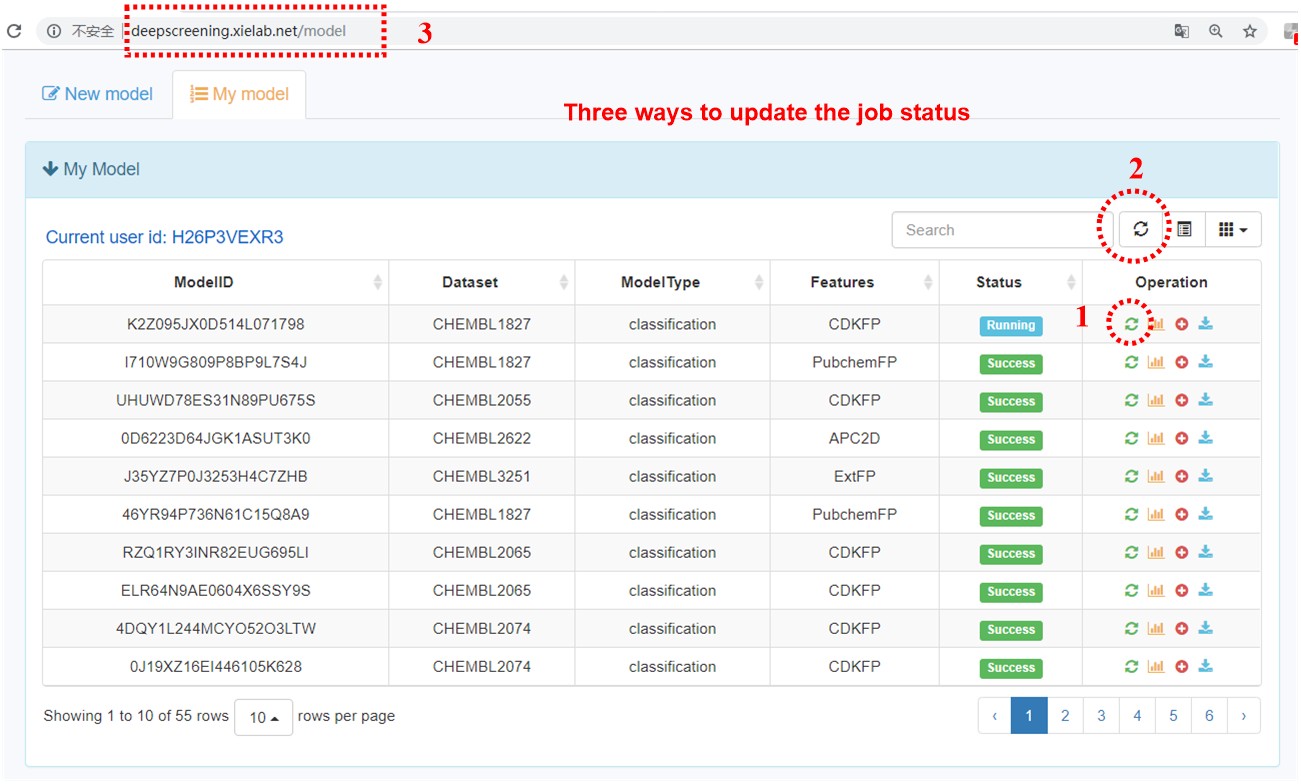
8. Three ways to update the job status.
After submitting a model, the status will be assigned “Running”. Approach 1: User can click the first button of “Operation” column of the table. Approach 2: User can also update the table through click the refresh button at the upright corner. Approach 3: Users could refresh the page by click the refresh buffer.
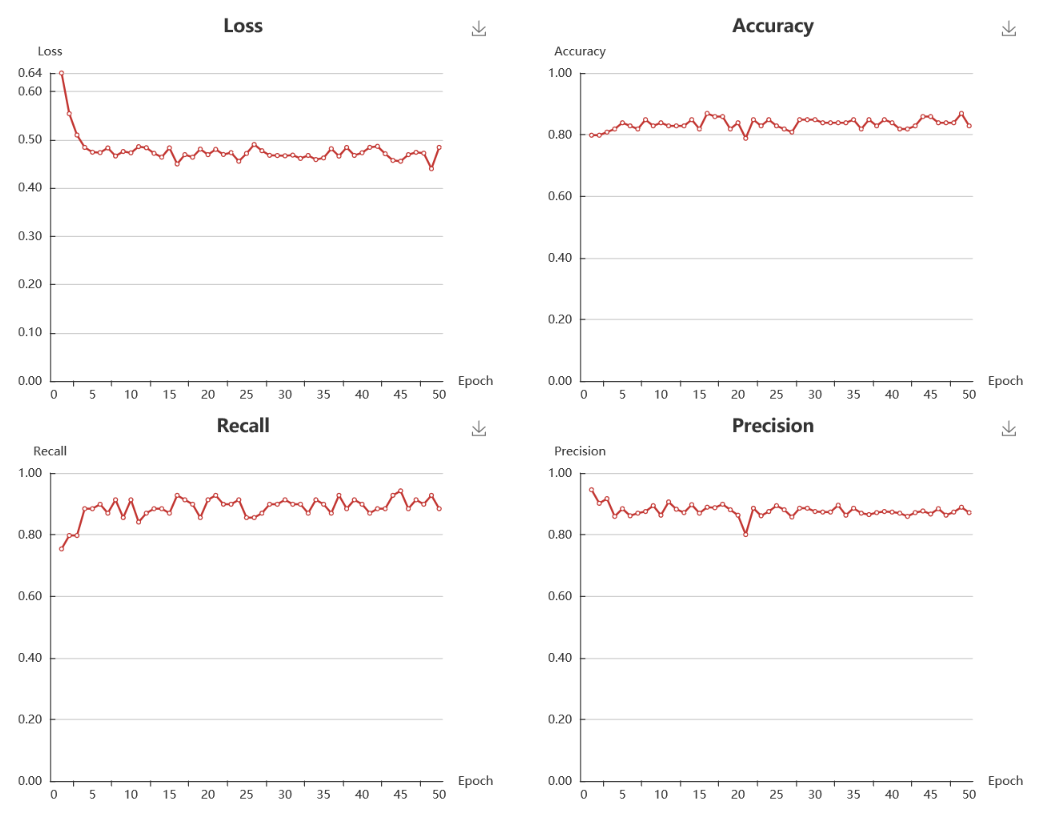
9. Metrics to evaluate the performance of a model.
For classification model, the test loss, accuracy, recall, precision, F1, and MCC are provided. All of those metrics are ranging from 0 to 1, and a greater value indicating a better model except for the test loss. The detail calculation formula can be further explored in https://en.wikipedia.org/wiki/Confusion_matrix. For regression model, the test loss, R2, MSE, RMSE, and AME are provided.
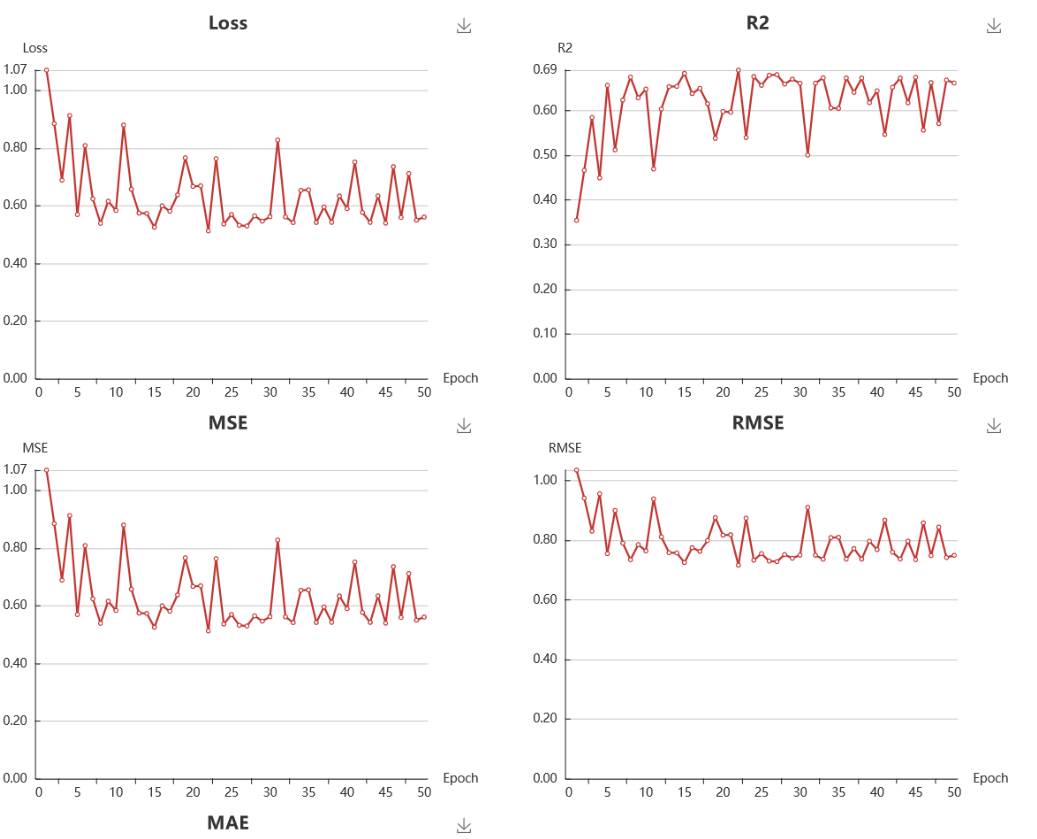
10. Ways to improve the model.
We encourage users to change parameters and try different fingerprints to improve the model performance when model is not as good as expected using the default parameters.
11. Steps to virtual screening a library using constructed deep learning models.
Step 1: Select a constructed model from model list or click the screening button from model list.
Step 2: Upload a chemical library with SDF file format, or select a chemical library from table. DeepScreening has prepared a in stock diverse library covering synthetic compounds, natural products, drugs, covalent agents, PPI and allosteric modulators collected from various chemical vendors. The fingerprints of those libraries have been precalculated and are ready to screening.
Step 3: Click the submit button and turn to the My screening page to check the job status.

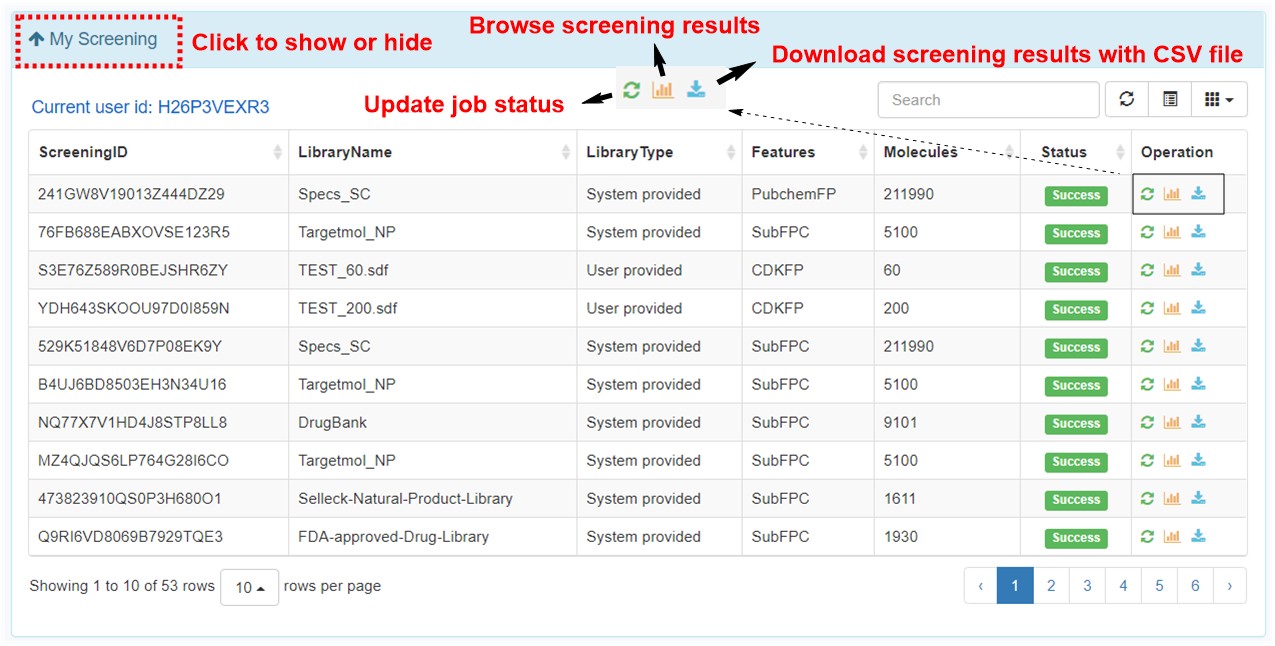
12. Functions in my screening page.
In my screening page, user can browse the screening jobs in an interactive table. When job is finished, user can click the browse button to assess the screening results, and click the download button to download the prediction results in a csv file.
13. Interpretation of the screening results.
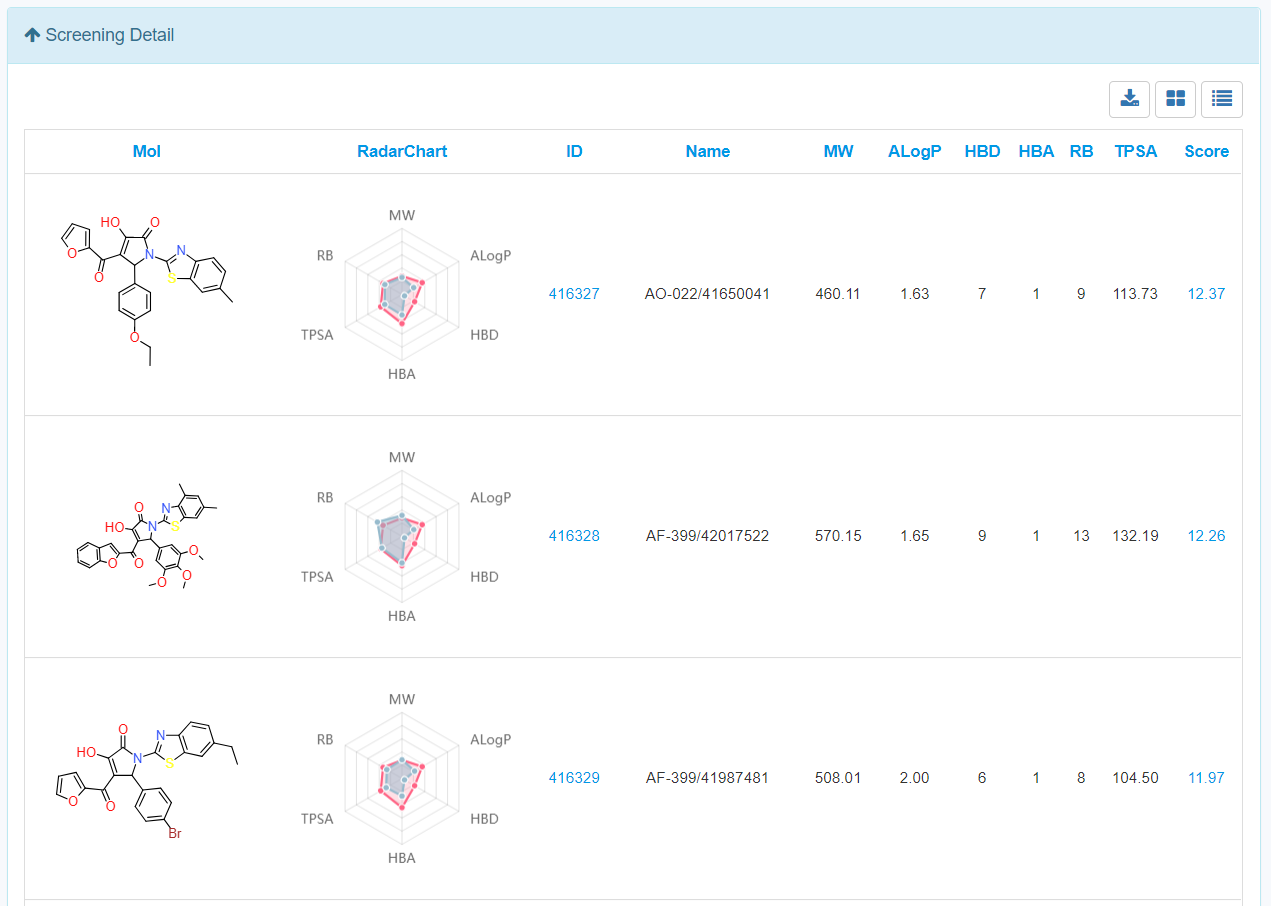
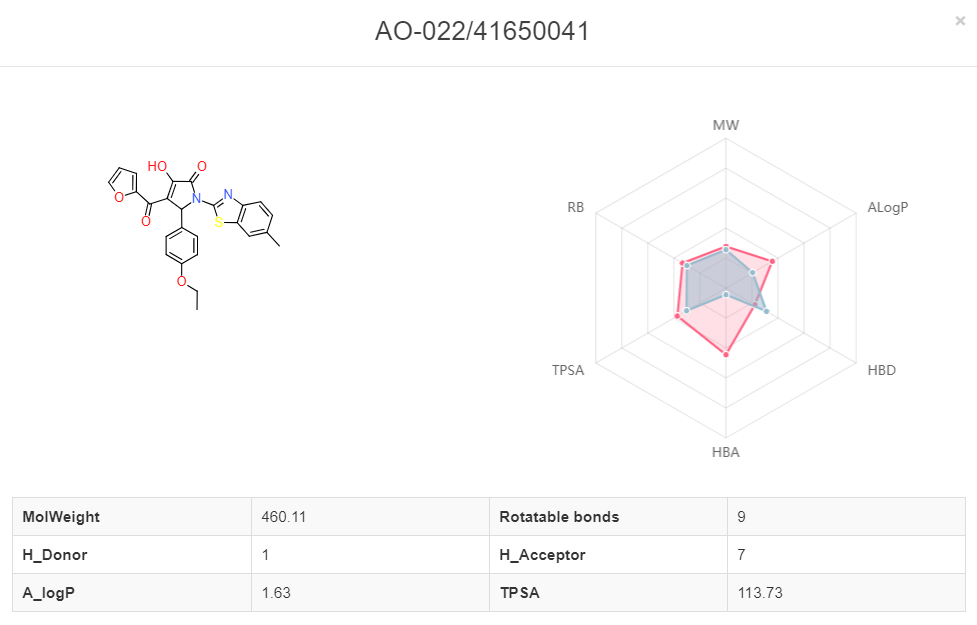
Click the screening detail button in my screening page, and turn to the “Screening Detail” section as shown below. There are grid view and table view for screening hits. The Compound ID is located at up-left, and the predicted score is located at down-left. For classification models, the scores are the probabilities (0~1). For regression models, the scores are the pIC50, pEC50, pKi, pKd. Greater score indicates a better affinity. Click the plus button to check the radar plot of the drug-like properties. Red present the desired drug-like properties (MW: 500 g/mol, ALogP: 5, HBA: 10, HBD: 5, TPSA: 140, RB: 10) according to the Lipinkski's rule of 5.Click the up-right download button to download the SDF files with a maximal of 500 compounds.

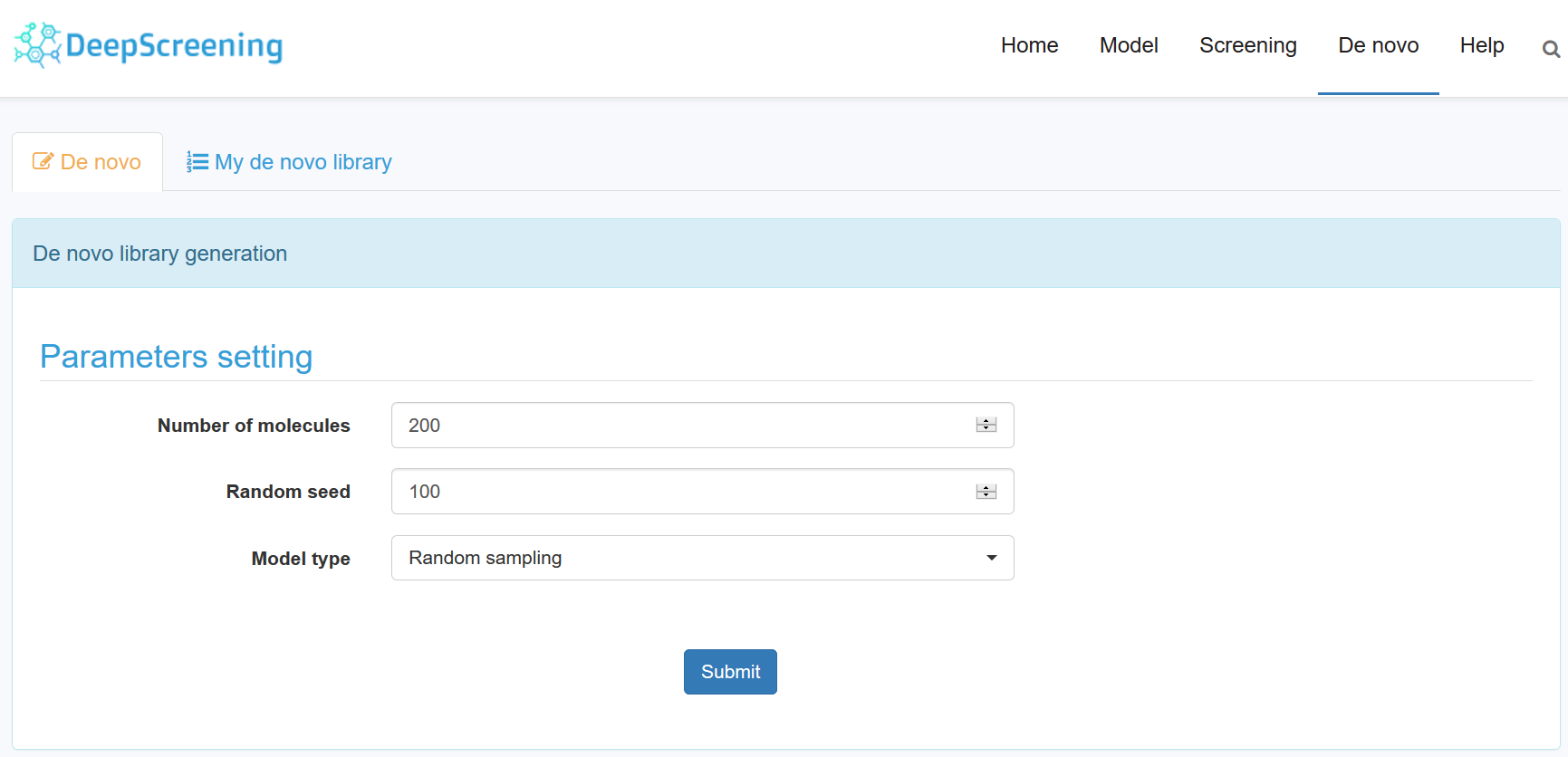
14. Generate a de novo library.
Click the “De novo” link in the home page. The de novo library generation is based on the pretrained RNN model on more than one million structures (using smiles as formats) from ChEMBL. Furthermore, user can generate a target focused library through transfer learning on a new targeted dataset, which can be select from ChEMBL or provided by users. The RNN based de novo library generation framework used in DeepScreening was based on REINVENT and QBMG. Their pioneering work has demonstrated that RNN is a powerful tool for de novo drug design. The details can be seen in the original references (Olivecrona, M., et al. J Cheminform (2017) 9: 48. Zheng, S., et al. (2019), J Cheminform, 11 (1), 5).
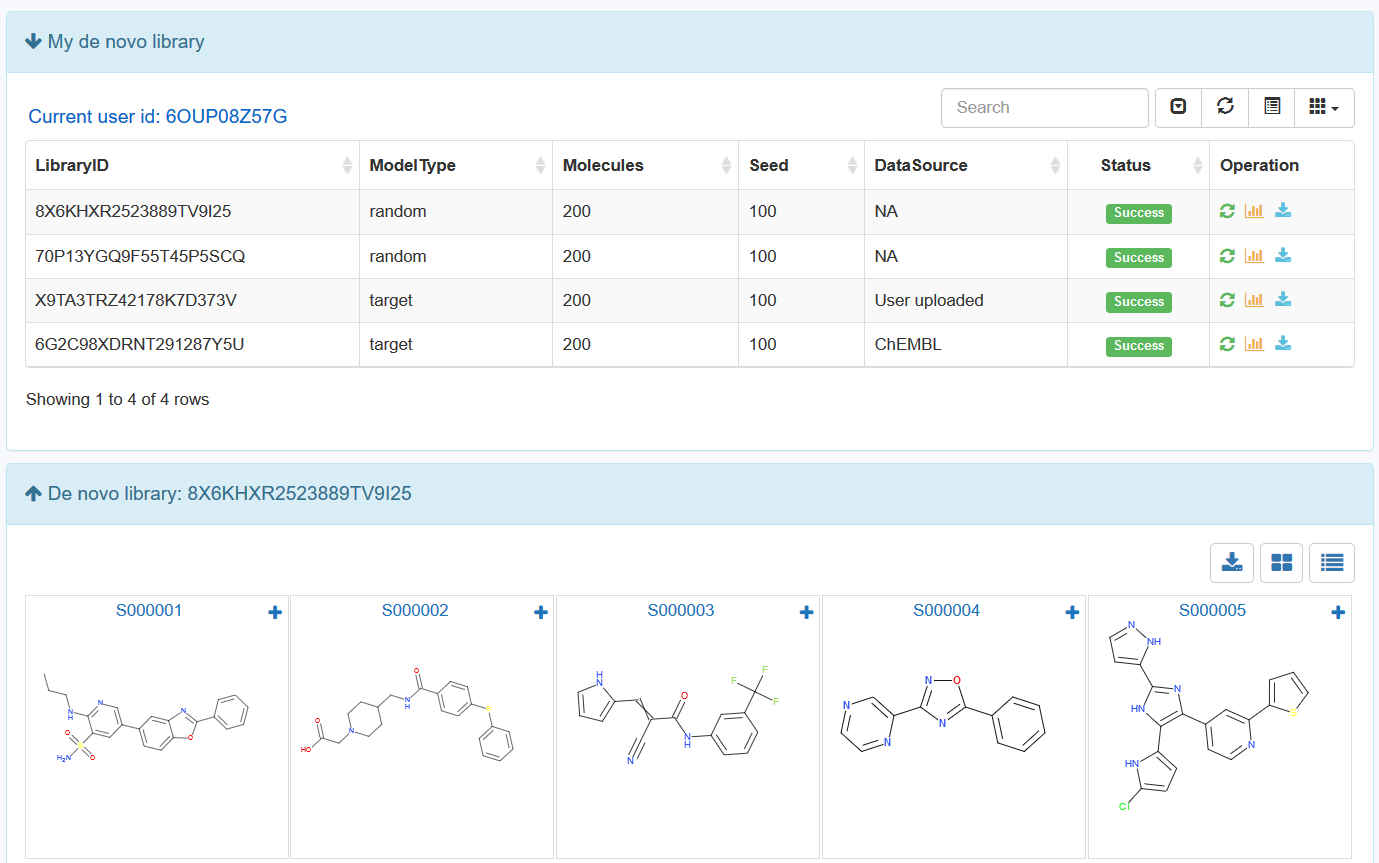
15. Functions in “My de novo library” page.
After submitting a de novo task, user can browse all the de novo libraries in an interactive table with searching function. In the operation filed, user can update job status. When the job is finished with a status sign of “Success”, user can browse the library details, and download the library to local computer.
About us
Research Group for Gut Microbiome and Health (RGGMH) at Guangdong Institute of Microbiology was established by Dr. Liwei Xie and funded by grants from Guangdong Academy of Sciences and Guangdong Institute of Microbiology. Dr. Liwei Xie was trained in the area of both Molecular Nutrition and Computational Biology in United States in 2007-2017. The research group focuses on the AI-algorithm development and application in drug design, functional and health benefit small molecules and nature products design and screening, targeting gut microbial dysbiosis-associated metabolic diseases, such as Type 2 diabetes (T2D) and obesity. Our group has state of art research facilities including high-throughput screening and computation system, single cell screening, isolation and sequencing system, health and disease related transgenic mice and gut microbiome models. We aim to provide basic biomedical training not only for graduate students and postdoctoral researchers, but also for clinical students and doctors with respect to the translational studies.
Address
State Key Laboratory of Applied Microbiology Southern China
Guangdong Institute of Microbiology
100 Xianlie Middle Rd, BLD58 2rd Floor
Guangzhou, Guangdong, China, 510070
Contact
Telephone :+86-020-87137679
Fax: +86-020-37656322
Cell: +86-17688872852
Email: xielw@gdim.cn
Citing us
Liu, Z.; Du, J.; Fang, J.; Yin, Y.; Xu, G.; Xie, L. DeepScreening: A Deep Learning-Based Screening Web Server for Accelerating Drug Discovery. Database 2019, 2019, 1–11. https://doi.org/10.1093/database/baz104.